Playlist
Show Playlist
Hide Playlist
DNA Repair Defects & Neoplastic Molecular Markers – Carcinogenesis

-
Slides Carcinogenesis Basic Principles.pdf
-
Download Lecture Overview
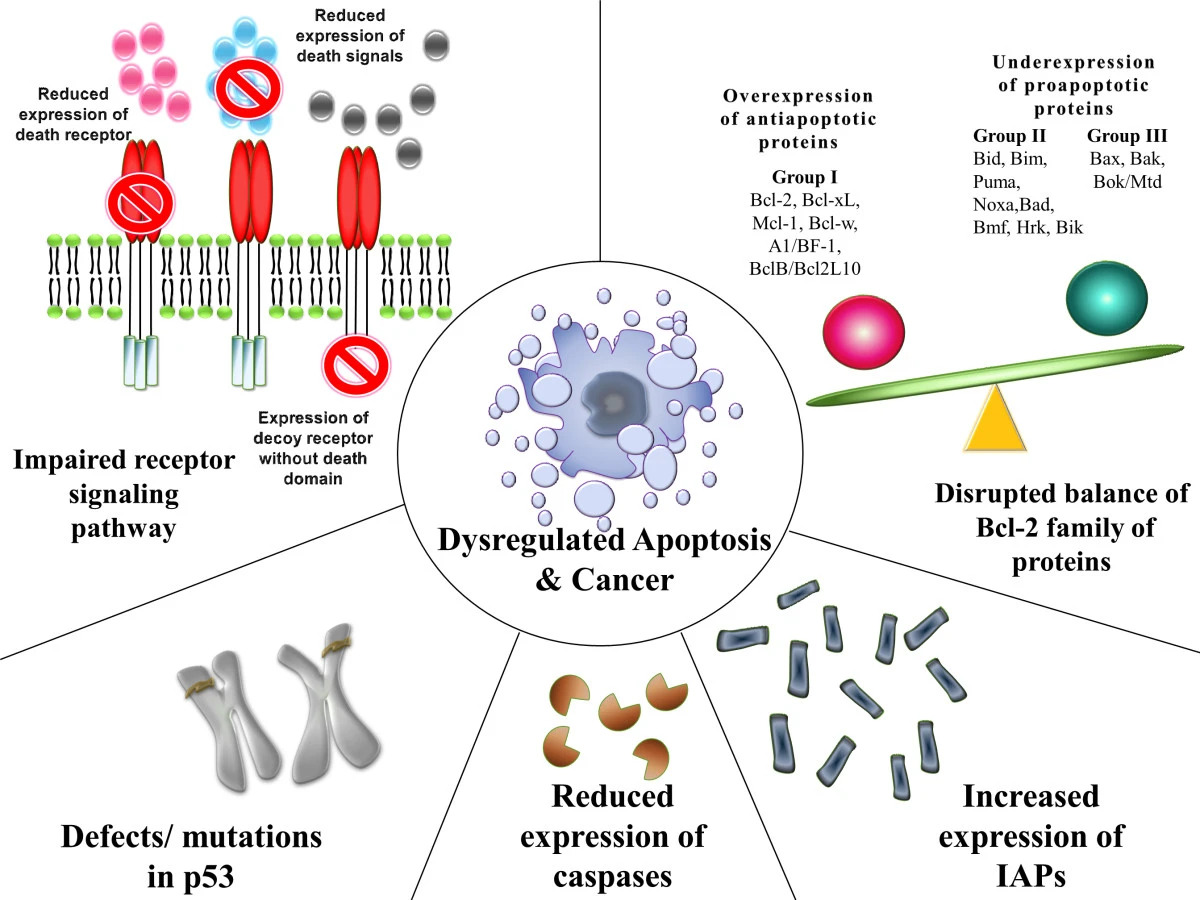
00:01 You'll notice here, on the flow chart thus far, we've walked through our box on the left in which we talked about chemicals, viruses and radiation. When a normal cell has been exposed increases the risk of cancer. 00:19 Our next topic, which is a very short topic is that if there is failure of your DNA repair which we already kind of discussed with xeroderma pigmentosa. 00:29 Upon exposure to UVB rays may then develop cancer. DNA repair defects. Let's talk about this in great detail. 00:40 What you find here on the right, is the fact that you have your DNA, and you have a double helix. 00:48 Next, you find depurination and then you have a, what's known as a endonuclease. And then finally DNA polymerase in which it then helps you put back a part of the strand which is then been removed. You'll notice here please, that on the bottom strip, from 5' to 3', that you have a nucleotide endonuclease taking out the G. 01:18 And then you have a polymerase and along with the ligase which puts it all together. 01:23 Now what ends up happening on the right, is the fact that you are not able to properly do it. Deaminase. 01:29 Then you have DNA glycosylase. Then you have endonuclease and DNA polymerase. So these are the enzymes that you want to know, in general from genetics. I'm not going to go into greater detail about this apart from the fact that some of this enzymes may then become mutated. Mistakes made in DNA replication are corrected by DNA repair genes usually. 01:49 Often detected by what's known as microsatellite instability. What is a microsatellite instability and why do you want to know this. If you are not able to properly remove a microsatellite instability, you might develop a condition known as, HNPCC, hereditary non-polyposis colorectal cancer. This may then give rise to a right sided colorectal cancer. 02:16 Clinical application of what we are looking at here, i'm not giving you information that is trivial. 02:22 Every single point has serious clinical significance. So, what then happens is that if there is a mutation in the DNA repair enzymes, then this then becomes fixed. What becomes fixed? The microsatellite. What's a microsatellite? The 1 to 6 nucleotide tandems that should normally be removed by the enzymes. The mutation is in one of the genes. 02:47 At least want to know MSH 2, MLH 1. Memorise that. The mutations in MSH and MLH result in a condition called Lynch syndrome. 03:01 What is another name for Lynch? Hereditary non-polyposis colorectal cancer. Giving rise to what kind of colorectal cancer? More so right of left side? Right. Another condition which we talked about earlier with DNA repair mutations, is your thymine dimer, with xeroderma pigmentosa. Walked through this in greater detail earlier. 03:27 Upon exposure to UVB rays, may develop certain types of skin cancers. Be familiar with the enzymes that we are seing here on the right and what it's responsible for doing in terms of proper replacement Our next big topic is the fact that we are going to take each one of this boxes, i'm going to first read them through you or to you and then we'll walk through them to show you that if you are able to satisfy each one of the criteria in this boxes, what are you doing? You are developing cancer. 04:07 The first box on your left is oncogenes. If i gave you t(8;14), you tell me, Burkitt. And you would tell me what kind of oncogene? C-myc. Lots of c-myc, gives rise to cancer. What kind? Burkitt. The middle box. 04:28 Remember the tumor suppressor genes are security guards. They are the guardians of your cell cycle. 04:36 As you walk through your cell cycle, our major point of arrest would be between G1 and S phase. 04:41 Remember G1 and S. Your focus on your boards will be between G1 and S. The guardians for G1 to S will be Rb and p53. If the guardians have been removed for whatever reason. p53 being very common. It is the most common mutated gene isn't it. And if p53 has been removed there is nothing stopping the cell from going to a cell cycle. 05:10 We'll talk about this in greater detail. And finally we will talk about apoptosis in which if a cell is able to successfully evade apoptosis, then the cancer cell is never going to die. Clear? Each one of this boxes, clinically significant. 05:29 Talk about the types of mutation here. We will have to go through in great detail, RAS. You have heard of KRAS, NRAS, HRAS. 05:39 Our focus will be KRAS. You have heard of TP53? And actually from henceforth, whenever you see the letter 'P', in front of a number and such, that to you should indicate that tumor suppressor gene. And we have RB. RB being another big point of mutation Translocations (9;22), well apart from CML I want to add another cancer that you need to know. 06:05 Would you tell me what is the most common leukaemia in a child between the ages of 1 to approximately 15? Acute Lymphoblastic Leukaemia. t(8;14) Burkitt, t(14;18) Good. Follicular. t(15;17) AML type 3. We will talk about this later. 06:24 Deletions, amplifications, ERBB2. ERBB2 is synonymous with HER2/neu. So what we will do, and don't memorise this right now. 06:37 I'm just giving you types of mutations that are to come. And they will come. And we will keep repeating, reinforcing you will be sick and tired of me if you are not already, of all the different mutations that we have to go through including BCL2. 06:55 The genes involved in cancer. Proto-oncogenes we will talk about. Remember, proto-oncogenes are actually normal. 07:02 We require for proper growth. If then these become mutated this then become oncogenes. The suppressor genes, p53 and RB. 07:12 Anti-apoptotic gene will be BCL2. And we already did our DNA repair gene in which we had mismatch repair and we talked about MLH1 and MSH2. And we will talk about apoptosis as well. Must know. Either between BCL2 and BAX. 07:28 Nice little introduction of high yield list of genes and mutations that you have to know for your boards. 07:37 The topic on this table. These tables are big time. Neoplastic molecular markers and when do you want to use it. 07:45 Let's first begin with the first row and with ABL I want you to jump over to t(9;22). So whenever you hear t(9;22) you should be thinking about Philadelphia chromosome. That t(9;22) that translocation gives rise to and codes for an enzyme called tyrosine kinase. And whenever this translocation takes place you will automatically have CML. 08:08 And if it's a young patient I was talking to you about leukaemia, and then it would be ALL. 08:13 So what's a non-receptor TK activity. TK stands for tyrosine kinase. In pharmacology, you have learned of a drug in which it knocks out the tyrosine kinase activity of t(9;22). Welcome to imatinib. Next, we have HER. 08:29 HER. Now interesting enough the marker gives you the gender. So it's a she. Now with this gender, what does HER actually stands for? Human epidermal growth factor receptor. Epidermal growth factor receptor. 08:44 So what does that mean. It means that the receptor autonomously undergoes amplification, and with all this receptor activity what kind of cancer is your patient going to develop? Breast cancer. 08:56 And this type of breast cancer with HER2/neu positive, you are going to use a drug called? Good. Trastuzumab. 09:05 MYC. With MYC it's a nuclear transcription t(8;14) Burkitt. So down in the area of the nucleus, inside the nucleus, you have an oncogene, specifically focusing upon C-myc and this then gives rise to Burkitt. 09:20 Then you have something called N-myc. N-myc. N as in N-myc, N as in neuroblastoma. Also responsible for nuclear transcription. 09:32 We will be spending quite a bit of time with RAS. You need to know everything about RAS. 40-50% of your cancers has a RAS mutation involved. At this point, I would like for you to connect and forever memorise the RAS associated with GTP signal transduction. GTP, RAS. When we get into GI, we will talk about what's known as familial adenomatous polyposis. With familial adenomatous polyposis there is a gene, a mutation with APC. 10:07 At some point, I will give you the interaction between APC and beta catenin. If this then becomes mutated, what's your risk of going on to colorectal cancer in familial adenomatoud polyposis? 100%. What are you going to find in the colon? A carpet of polyps. And associations of familial adenomatous polyposis, might have heard of Gardner and you know about the Turcot. 10:35 Next we have BRCA1 and BRCA2. BRCA, breast cancer. Not only could it be breast, it could also be the ovary. 10:45 Both will be involved. One and two. RB. Retinoblastoma. This is a normal tumor suppressor gene, that guards a cell or guards, monitors, supervises the quality of a cell as it goes from G1 to S phase. 11:05 G1 to S phase, that is perfectly normal. If there is a mutation in chromosome 13 and retinoblastoma has been removed, you remove the break. Worried about retinoblastoma, and later on in life genetically may develop osteosarcoma. 11:24 TP53 is all over the place. Also controls and monitors the quality of your cell between G1 and S. 11:32 If you have'nt already, you put together p53 and RB they will work together. If those are become mutated, these then give rise to all kinds of cancer. p53 is the most common mutation that results in cancer development.
About the Lecture
The lecture DNA Repair Defects & Neoplastic Molecular Markers – Carcinogenesis by Carlo Raj, MD is from the course Cellular Pathology: Basic Principles with Carlo Raj.
Included Quiz Questions
A mistake in which of the following enzymes would NOT result in DNA repair defects?
- Deiodinase
- Deaminase
- DNA glycosylase
- Endonuclease
- DNA polymerase
Which of the following are associated with hereditary nonpolyposis cancer syndrome?
- MSH2 and MLH1 mutations
- Thymine dimer mutation
- t(8:14)
- C-myc mutation
- Rb mutation
Which of the following is NOT a proto-oncogene involved in carcinogenesis?
- BAX
- ERBB2
- RAS
- ABL
- MYC
Which is the translocation associated with CML termed the Philadelphia chromosome?
- t(9;22)
- t(8;14)
- t(15;17)
- t(14;18)
- t(9;12)
Which of the following molecular markers is associated with 50% of all cancer diagnoses?
- RAS
- ERBB2
- N-MYC
- MYC
- ABL
Author of lecture DNA Repair Defects & Neoplastic Molecular Markers – Carcinogenesis

Carlo Raj, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
1 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
I absolutely love this lecturer. He goes straight to the point, gives high-yield and applicable information (lots of information btw) in a summarized awesome way. 5 starts no doubt.
