Playlist
Show Playlist
Hide Playlist
Cystic Fibrosis (CF): Diagnosis & Management

-
Slides Cystic Fibrosis.pdf
-
Download Lecture Overview
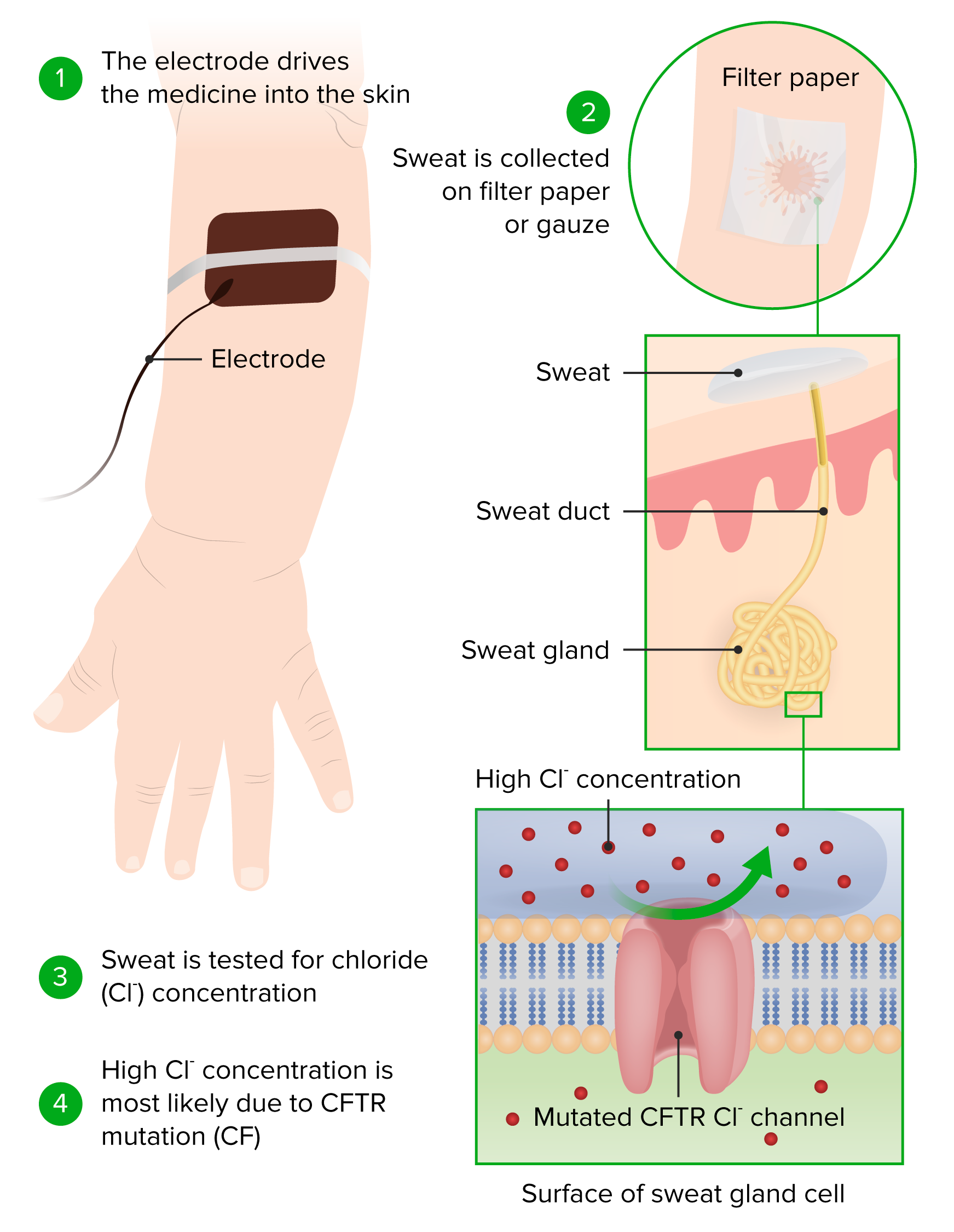
00:01 So if we suspect cystic fibrosis, how do we diagnose the disease? Well, the first test we should go to is actually quite inexpensive and it’s the sweat test. 00:14 Typically, we take a child, we put them under a hot lamp for an hour, and we allow them to sweat. 00:22 And we have patches that are on their skin and we weigh the patches and then we measure the salinity of the patch. 00:29 That will tell us whether they have cystic fibrosis by virtue of the fact that children with chloride channel defects will have abnormal salt content in their sweat. 00:42 If we then suspect CF in this patient, oftentimes we’ll do a DNA analysis and find what are the mutations that are present in this child’s CFTR gene. 00:54 So when do you want to get a test? You certainly want to get a test in a child with recurrent cough or pneumonia, a child with recurrent sinusitis, a child with poor weight gain or failure to thrive, a child with rectal prolapse or nasal polyps, any child where there’s a family history of cystic fibrosis, a child with a fat soluble vitamin deficiency, a child with clubbing, and certainly a child with Pseudomonas or other atypical lung infections. 01:23 In all of these cases a sweat test is indicated. 01:27 Remember, it’s a cheap and accurate test. 01:32 Treatment of these patients is complex and we’ll break it down for you. 01:37 First off, antibiotics are really indicated for all respiratory exacerbations. 01:42 Chances are this patient has an overgrowth of bacteria. 01:47 The goal here is suppression of the growth of bacteria, it’s not a cure. 01:52 We can’t truly eliminate these bacteria, especially the ones growing in biofilms. 01:58 Long-term, patients will get monthly tobramycin inhalational therapy or they may also get azithromycin. 02:07 Keep in mind though the azithromycin is mostly for its anti-inflammatory properties, not for its antimicrobial properties. 02:17 Other inhaled medications include nebulized 7% saline. 02:23 This acts as an osmotic agent and allows fluid to flow into the airway which loosens up the mucus. 02:30 These patients will require a pulmonary toilet, which is all the therapies that go with this, such as coughalators and vest treatments. 02:39 Patients will often get DNAses, which they have to then inhale. 02:44 These are enzymes that cleave long strands of neutrophil DNA An example of a name is Pulmozyme. 02:51 This thins out the mucus further to allow these patients to cough it up. 02:56 They usually have asthma along with this so they are often treated with albuterol and steroids, as we do with asthma. 03:03 And these patients, we need to make sure they get immunized. 03:07 Influenza can really throw a patient with cystic fibrosis for a loop. 03:12 The flu vaccine is very important, so is the streptococcal infection vaccines like the multivalent streptococcal infection that we give in infancy or even the Pneumovax vaccine. 03:27 For severe disease, these patients may end up on BiPAP, especially later on in life. 03:32 As a continuous therapy they may have a ventilator machine at home. 03:35 As a continuous therapy they may have a ventilator machine at home. 03:35 CFTR modulators are a class of drugs that target the production, processing, or function of defective CFTR protein, and are associated with improved lung function, improved quality of life, and a reduction in exacerbations. 03:52 Several options are available, and are chosen based on the specific CFTR gene mutation present. 04:01 They may end up getting embolization or surgery for any pulmonary hemorrhages that happen. 04:06 And in very last case, we may treat them with lung transplant. 04:12 If you are growing Burkholderia, you probably are not allowed to be given a lung transplant, you’re excluded from the list. 04:21 Lungs transplant is not taken lightly. 04:25 The five-year survival rate of a patient with lung transplant is around 50%. 04:32 Other treatment is required for other presentations of the illness. 04:37 For sinusitis, patients will get antibiotics, nasal steroids, rarely surgery. 04:42 For pancreatic disease, we give them extra fat soluble vitamins and insulin if they have the associated diabetes. 04:50 For biliary disease, we will give these patients ursodeoxycholic acid, which dissolves gallstones. 04:57 And if they have very severe liver disease, they may require a liver transplant. 05:02 And for the distal intestinal disease like DIOS, we’ll reduce any rectal prolapse that happens to come along, and what’s key is making sure we avoid constipation with meds like polyethylene glycol. 05:18 The prognosis is dim for these families and for these patients. 05:22 Nearly every patient with cystic fibrosis will pass away prematurely. 05:27 But the age at which people are surviving is later and later and some people are living well into their parenthood and are able to have outstanding and productive lives. 05:38 So this is a disease that we are improving how patients are doing. 05:46 Some key aspects of better diagnosis early is resulting in a better outcome. 05:54 So it’s key to remember that we have to diagnose these children early in their childhood and that will confer a longer and more healthy life. 06:03 We have to counsel patients at all stages of life in terms of what their expectations are and in terms of what their needs are. 06:12 It’s been shown that a center of care for cystic fibrosis management confers a better outcome than an outlying area. 06:20 So patients should be referred to a major CF center where they can get a cutting-edge care. 06:27 That’s all I have for you today about this illness. 06:29 Thanks for listening.
About the Lecture
The lecture Cystic Fibrosis (CF): Diagnosis & Management by Brian Alverson, MD is from the course Pediatric Pulmonology. It contains the following chapters:
- Diagnosing the Disease
- Respiratory Treatment
- Other Treatment
- Prognosis
Included Quiz Questions
What is the investigation of choice for a child where we suspect cystic fibrosis?
- Sweat chloride test
- Complete blood count
- Lipase and liver function testing
- Basic metabolic panel/Chem 7
- Bronchoscopy
A child is about to undergo a sweat chloride test for suspected Cystic Fibrosis. All of the following are an indication of this test, EXCEPT?
- Recurrent meningitis
- Recurrent Sinusitis
- Recurrent pneumonia
- Failure to thrive
- Rectal prolapse
Which of the following histological abnormality is expressed in sweat testing?
- Chloride channel defect.
- Sodium channel defect.
- Potassium channel defect.
- Calcium channel defect.
- Phosphate channel defect.
Author of lecture Cystic Fibrosis (CF): Diagnosis & Management

Brian Alverson, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
1 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
This topic deserves a series of lecture due to its multi-systemic nature. Thank you!
