Playlist
Show Playlist
Hide Playlist
Congenital Adrenal Hyperplasia (CAH)

-
Slides Reproductive Endocrine Disorders.pdf
-
Reference List Endocrinology.pdf
-
Reference List Reproductive Endocrine Disorders.pdf
-
Download Lecture Overview
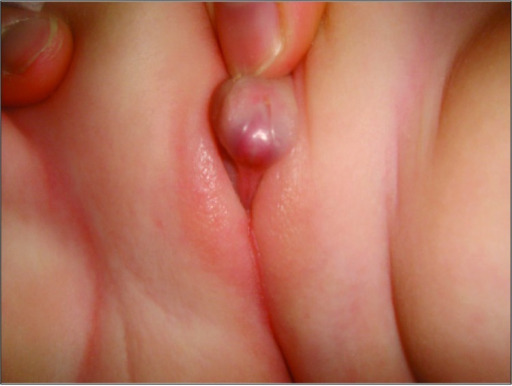
00:01 Congenital adrenal hyperplasia is a group of autosomal recessive enzyme deficiencies that are characterized by impaired normal corticosteroid synthesis in the zona fasciculata of the adrenal cortex 21-hydroxylase deficiency is by far the most common enzyme deficiency leading to congenital adrenal hyperplasia in over 90% of cases. 00:23 This is then followed by the much rarer 11-beta hydroxylase deficiency and 17-alpha hydroxylase deficiency Classical salt-wasting congenital adrenal hyperplasia is the most severe form of the condition. 00:39 The clinical features of this condition include manifestations of hyperandrogenism. 00:45 At birth, there is ambiguous genitalia, enlarged clitoris and fused labia in females. 00:51 Small testes in comparison to the phallus and scrotal hyperpigmentation in male. 00:56 In children, precocius puberty tend to be tall, patients tend to be tall with an increased bone age but short adults due to premature closure of the epiphysis due to absent hormonal stimulation. 01:09 In adolescents and adulthood, temporal balding, acne, irregular menses, hirsutism, polycystic ovaries and infertility may be found. 01:20 The manifestations of mineralocorticoid deficiency of this condition include renal salt wasting or excessive sodium secreted in the urine. 01:30 Symptoms at birth will include poor feeding, weight loss, vomiting, dehydration, low blood pressure and alterations in potassium and sodium as well as metabolic acidosis. 01:42 Many of these were present in our case. 01:46 the occurence of adrenal crisis as early as 1-4 weeks of age, can manifest with azotemia, vascular collapse, shock and death. 01:56 That's why it is imperative to make the diagnosis of the condition and begin treatment immediately. 02:02 In the classic form of congenital adrenal hyperplasia, the level of 17-hydroxyprogesterone is increased. 02:10 In the non-classical form, 17-hydroxyprogesterone levels are not as high as in the classical form. 02:16 Adrenocorticotrophic hormone stimulation test as we performed on this patient should be done if medical history, family history, and physical examination suggest the 21-hydroxylase deficiency With this test, 250 micrograms of ACTH is given intravenously and 17-hydroxyprogesterone levels are measured at baseline and then after one hour The test can be repeated if the initial results are negative. 02:44 Plasma renin activity is inversely related to intravascular volume status and serum sodium. 02:50 In classical salt wasting congenital adrenal hyperplasia, these values are increased. 02:57 In patients with a strong family history of congenital adrenal hyperplasia, genetic analysis is recommended. 03:04 If both parents are carriers, prenatal treatment and antenatal treatment with dexamethasone is recommended until genetic diagnosis of the fetus can be performed by chorionic villus sampling or amniocentesis. 03:19 Here is a table that goes through comparative features of the various forms of congenital adrenal hyperplasia By far the most common one is the 21-hydroxylase deficiency which occurs in over 90% of cases. 03:34 In this situation, patients will manifest with androgenization and decreased mineralicorticoid effects In the 11-beta hydroxylase deficiency which occurs in 5% of patients, there will be androgenization but mineralocorticoid activity will be increased. 03:53 And then finally, in the much rarer 17-alpha hydroxylase deficiency, the adrogenization is minimized and the mineralocorticoid effects are prominent.
About the Lecture
The lecture Congenital Adrenal Hyperplasia (CAH) by Michael Lazarus, MD is from the course Reproductive Endocrine Disorders. It contains the following chapters:
- Congenital Adrenal Hyperplasia (CAH)
- Enzyme Deficiencies Resulting in CAH
Included Quiz Questions
Which enzyme deficiency is most commonly associated with congenital adrenal hyperplasia?
- 21-hydroxylase deficiency
- Prolyl 3-hydroxylase deficiency
- 11-beta-hydroxylase deficiency
- Lysyl hydroxylase deficiency
- 17-alpha-hydroxylase deficiency
Which of the following findings is most likely in a patient with a mineralocorticoid deficiency?
- Renal salt wasting
- Small gonads
- Amenorrhea
- Short stature
- Polycystic ovaries
What is the preferred prenatal treatment for fetuses with two parents who are genetic carriers for CAH?
- Dexamethasone
- Testosterone
- Estrogen
- Aldosterone
- Sodium supplementation
Author of lecture Congenital Adrenal Hyperplasia (CAH)

Michael Lazarus, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
