Playlist
Show Playlist
Hide Playlist
Congenital Adrenal Hyperplasia (CAH): Diagnosis, Treatment, Outcomes and Infertility

-
Slides Congenital Adrenal Hyperplasia.pdf
-
Download Lecture Overview
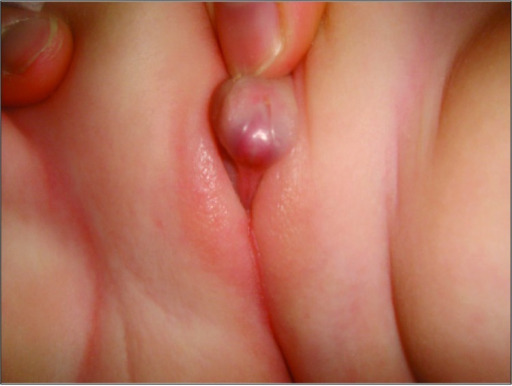
00:01 Let’s now review classical versus non-classical 21 hydroxylase deficiency. 00:07 In classical deficiencies, there is complete enzymatic deficiency. 00:11 There is either a frameshift or nonsense mutation that causes the enzyme to completely not function. 00:18 You have prenatal masculinization at birth, with low cortisol, elevated ACTH and 75% of these patients will have salt wasting which is life threatening. 00:32 With non-classical, it usually presents later on in life typically at puberty or in the reproductive time. 00:40 This is a mild enzymatic deficiency so you may have a nucleotide substitution. 00:45 This is a gradual postnatal development with high androgens or hyperandrogenism with variable virilization across the spectrum. 00:55 These patients typically have normal cortisol and normal ACTH. 01:00 They do not have the salt wasting phenomenon that you see with classical 21 hydroxylase deficiency. 01:07 Let’s then review a more experimental protocol. 01:09 I doubt that you'll have any exam questions regarding this but just so you know, if you'd like more details, I’ll explain it here. If not, feel free to move on to the next set of slides. 01:19 Here, in our prenatal treatment algorithm consists of one confirming that there's a pregnancy in a patient that we suspect may pass on 21 hydroxylase deficiency. 01:30 We confirm pregnancy by doing an HCG or human Chorionic Gonadotropin protein analysis, then, once we've confirmed that there's pregnancy we start dexamethasone which is the steroid meant to suppress the adrenals and not only mom but also in the fetus as it crosses the placenta. 01:49 Then, what we’d like to do is establish a karyotype. 01:52 If it’s a male fetus, we don’t need to continue treatment so we do a karyotype through chorionic villi sampling which typically occurs between 9 and 11 weeks of gestational age then we’d complete a DNA analysis to determine if there is a deficiency. 02:08 If there is no deficiency we can go ahead and stop dexamethasone therapy. 02:13 And, again, if the fetus is xy, 46 xy, which is male, we can also stop the dexamethasone. 02:20 Once we've confirmed that it’s female then we would continue dexamethasone until we are able to ascertain if the mutation is present or not. 02:29 And then once the child is born, there needs to be some postnatal follow up. 02:34 So we’d like to know the birth weight, the length, the head circumference and developmental assessment and, again, if we suspect any 21 hydroxylase deficiency, a heel stick will be performed upon delivery which will tell you if there are elevated levels of 17-hydroxy progesterone. 02:52 The other thing that we can do in these babies after they're born is to do a 72 hour blood sample which will include electrolytes and plasma renin activity. 03:04 So let’s now talk about post natal diagnosis. 03:08 Again, I doubt that you'll have very many questions on this, but just in case, let’s follow here. 03:14 What we do is we do laboratory studies to confirm the diagnosis of 21 hydroxylase deficiency in an infant who displays ambiguous genetalia. 03:23 Remember that these babies are usually female karyotype 46 xx, but upon exam, what we’d like to know is whether they have a uterus which will help us to tell if there were sertoli cells ever present, and we’d like to know what their serum 17-hydroxy progesterone is. 03:42 Usually this is done with a heel stick among a whole other panel screening for the neonate. 03:49 This is an example of what ambiguous genitalia looks like upon examination. 03:56 So, after a baby is born and the baby is cleaned off and placed in the warmer, the first question the parents wanna know is it a boy or a girl. In this case, we cannot tell by phenotypic sex or by the genitalia, so in this case you'd like to tell parents and other family members we're not sure but you have a healthy baby, we need to do some more investigation. 04:20 Okay, now that you know what an ambiguous genitalia looks like, let’s talk about postnatal medical treatment. 04:26 Our goals are to normalize the androstenedione and testosterone levels. 04:31 We also need to replete the infant with glucocorticoids through hydrocortisone. 04:36 Mineralocorticoids also need to be replaced with 9 alpha fludrocortisone acetate. 04:42 Let’s now talk about postnatal surgical treatment. 04:45 In the past we’d use to do genitoplasty or surgery on the genetalia right after delivery to determine sex rearing. 04:54 Now, this has been recommended to be delayed later in life so that the child can be involved in their own health care. 05:01 Let’s talk about some of the procedures that are done though postnatally. 05:06 On occasion, the parents will insist that this done be postnatally and even later in life when it is done, these similar operations will be done to make the external genetalia appear more female. 05:19 The operation includes opening the vaginal introitus. The vaginal introitus is the entrance of the vagina. 05:26 Also, the urethral meatus is brought closer to the perineum as it sometimes can - you can sometimes find that urine actually exits through the clitoris. 05:38 Also, it’s important that we reduce the size of the clitoris although this is controversial because sometimes any type of operation or surgery on the clitoris can affect sensation which can lead to sexual dysfunction later in life. 05:54 The goals are though, to allow menstrual flow to occur, enable tampon use and vaginal intercourse, prevent urinary tract infections and preserve the glands and neurovascular bundle too minimize the loss of sensation. 06:11 Let’s talk now about the adult features of Congenital Adrenal Hyperplasia. 06:17 Remember, that they have hyperandrogenism so that means they have high circulating levels of androgen. 06:24 This can actually cause acne in 33% of all women who have Congenital Renal Hyperplasia. 06:31 They can also have alopecia which means hair loss at different stages. 06:37 This hair loss only refers to hair loss on the head because of the rest of their body actually has more hair than the general population. 06:46 This is called hirsutism. If you'd like to learn more about hirsutism, download the lecture regarding hirsutism. 06:52 This occurs in 59 - 78% of all women who have congenital adrenal hyperplasia. 06:59 They can also have premature pubarche which is early pubical hair or precocious puberty which is early puberty which is pathological. 07:09 They can also have amenorrhea which means never had any menstrual period or oligomenorrhea which means cycle lengths greater than 35 days that occurs in 54% of all women who have congenital adrenal hyperplasia. 07:23 They can also have chronic anovulation and PCOS or polycystic ovarian syndrome. 07:29 There's also a separate lecture regarding PCOS, if you'd like to download that to read more, but here in the ultrasound you can see a uterus that actually has a very thick endometrial lining, below the uterus you see here an ovary that has many follicles in a pearls in a string which is classical for polycystic ovarian syndrome. 07:51 Twelve percent of these woman actually experience infertility which is the inability to conceive in one year of unprotected intercourse. 07:59 These patients can also have severe dyspareunia. 08:03 Dyspareunia means pain during intercourse and they can have decrease or poor sexual satisfaction. 08:10 These patients unfortunately when they do become pregnant also have an increase risk of spontaneous abortion or miscarriage. 08:19 Let’s now talk about the gynecologic outcomes in Congenital Adrenal Hyperplasia. 08:26 Women who have classical CAH, Congenital Adrenal Hyperplasia, or non classical, NCAH often manifest with primary or secondary amenorrhea. 08:36 50% have PCOS again if you'd like to know more about PCOS please download the slide set regarding PCOS. 08:45 Both CAH and NCH women have infertility but NCAH women can conceive spontaneously more often. 08:53 Remember, they overall have a milder phynotype compared to classical CAH. 08:59 There’s still an increase risk of SAB or miscarriage prior to NCAH diagnosis. 09:05 Let’s now talk about the mechanism of infertility in these women who have congenital adrenal hyperplasia. 09:12 We believe that it’s the elevated progesterone concentrations which played an important role in preventing normal cyclicity or normal menstrual cycles. 09:21 Also progesterone can have an effect in the whole reproductive system. 09:28 It causes a thicker cervical mucus, they can actually have inadequate endometrial maturation and the endometrium is important for receiving the fertilized egg which becomes a blastocyst, the endometrial lining has to be just right so the blastocyst can implant. 09:47 And we know in this patients that they have impaired embryo implantation as well.
About the Lecture
The lecture Congenital Adrenal Hyperplasia (CAH): Diagnosis, Treatment, Outcomes and Infertility by Lynae Brayboy, MD is from the course Normal Puberty and Disorders of Sexual Development. It contains the following chapters:
- Prenatal and Postnatal Treatment
- Adult Clinical Features of CAH
- Gynecologic Outcomes and Mechanism of Infertility in CAH
Included Quiz Questions
What is the genetic alteration seen in non-classical 21-hydroxylase mild enzyme deficiency?
- Nucleotide substitution
- Frameshift
- Nonsense mutation
- Robertsonian translocation
- Reciprocal translocation
Which method is used to screen for 21-hydroxylase deficiency?
- Heel prick test
- Urinalysis for 21 hydroxylase deficiency
- Card test
- Serum estimation for 21 hydroxylase deficiency
- Whole blood estimation for 21 hydroxylase deficiency
What is NOT a part of the work-up for 21-hydroxylase deficiency?
- Heel prick test
- Post 72-hour blood sample
- Measurement of 17 hydroxylase deficiency
- Peripheral blood for DNA analysis
- Electrolytes and plasma renin activity assessment
Which of the following is NOT a sign or symptom of congenital adrenal hyperplasia?
- Normal menstrual cycle
- Precocious puberty
- Hirsutism
- Acne
- Infertility
Which of the following is a common gynecological outcome in girls with congenital adrenal hyperplasia?
- PCOS
- Serous cystadenoma
- Endometriosis
- Atrophic ovaries
- Tubal cysts
Which drug is given prenatally to girls with suspected 21-hydroxylase deficiency?
- Dexamethasone
- Antibiotics
- Rhogam
- Immunoglobulins
- Hormones
Author of lecture Congenital Adrenal Hyperplasia (CAH): Diagnosis, Treatment, Outcomes and Infertility

Lynae Brayboy, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
