Playlist
Show Playlist
Hide Playlist
Complement Mediated MPGN – Nephritic Syndrome

-
Slides Nephritic Syndrome.pdf
-
Reference List Nephrology.pdf
-
Download Lecture Overview
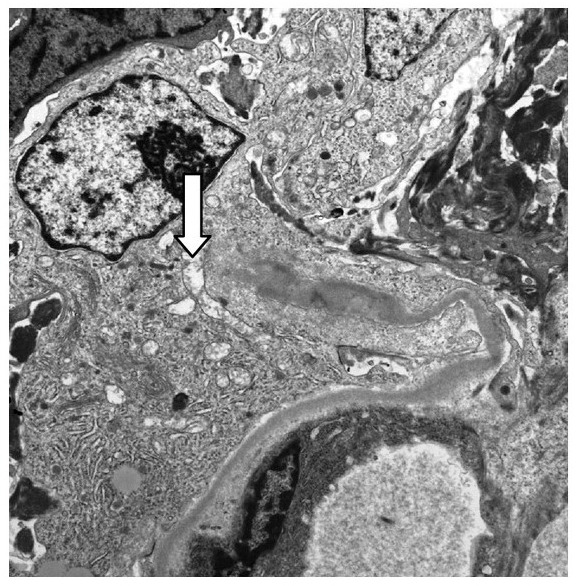
00:01 So, now we come to our complement-mediated MPGN. 00:05 We also refer to these as the C3 glomerulopathies. 00:09 There's two different types I want you to remember. 00:11 One, what we classically thought of as Type II MPGN called dense deposit disease or DDD. 00:18 The second is newly described C3GN and this has really been characterized since about 2010. 00:25 These are due to disorders of complement regulation. 00:31 So, that means that the activation or that there is activation of the alternative pathway if complement. 00:39 So, let's spend a moment talking about dense deposit disease. 00:42 It's relatively rare. It affects about 2-3 people per million and this is due to excessive activation of the alternative complement pathway as we talked about. 00:53 The clinical associations that you should remember in your DDD patients is that those patients can manifest with drusen. 01:01 Those are these areas in Bruch's membrane of the retina. 01:05 So, for anybody in ophthalmologist, it's very interesting to look at these retinal findings with an ophthalmologic or an ophthalmoscope. 01:12 Patients can also manifest with acquired partial lipodystrophy. 01:15 So, they may have this constitution together with their dense deposit disease. 01:19 This is really a disease of childhood. 01:22 So, my pediatric colleagues in nephrology are really gonna be seeing this more than I see it and by the time they come to me, they've typically already been transplanted. 01:32 So, when the pathogenesis of our complement mediated MGPN, so this is gonna include our C3GN and our dense deposit disease, there's a couple of different things that manifest in order to cause this injury. 01:43 Number one, patients can have a circulating autoantibody. 01:47 This is commonly referred to as C3 nephritic factor and it's present in about 70-80% of patients who have dense deposit disease. 01:56 Less so in our patients who have C3GN, about 40-50% of those patients. 02:01 You can also have -- or patients also have a deficient or mutated regulatory protein. 02:07 These are things that regulate that alternative pathway of complement activation and that is gonna include factor H and factor I. 02:16 So, when you have either that autoantibody that will turn on complement or you lose a regulatory factor that inhibits complement, the result is that you've got persistent activation and degradation of C3 and constitutive activation of alternative pathway. 02:36 So, again, if we focus on our complement pathway on the right which is our alternative pathway of complement activation, this is where my two factors are coming in. 02:47 If I've got C3 nephritic factor, that's that autoantibody that's circulating, that is going to turn on and constitutively, activate that C3 convertase, activating and using up our C3 so that's getting degraded, and then ultimately, activating our membrane attack complex. 03:05 If I have factor I that's diffecient or factor H, those are regulatory proteins that normally inhibit C3 convertase but if that is no longer there in the serum, again, constitutive activation of C3 convertase. 03:18 I've got C3 degradation, I've got turning on of that C5b-9 or membrane attack complex. 03:24 So, when we want to diagnose our patients or if we're thinking about that in our patient population, there's a couple of things that we can do. 03:33 We wanna look serologically at complement. 03:36 So, patients typically will have a low C3 complement. 03:40 This is because this is that alternative pathway of complement. 03:43 They're gonna have a normal C4, that's classical, so that's really an important way to distinguish whether somebody's got immune complex mediated or they're gonna have low C3 and C4, and just alternative pathway where they're gonna only have a low C3. 03:57 The AH50 which is alternative complement is also low in the serum. 04:01 So, again, when a patient has a low C3, activation of the alternative pathway, that really should prompt us as diagnosticians to further look at things that might be going on to disregulate that complement system. 04:15 We need to look for whether or not that patient might have mutations or allelic variants of complement factors. 04:21 And we can also do autoantibody assays to make sure that we don't have something like C3 nephritic factor and then look for those regulatory factors like factor H, factor I, again, C3 nephritic factor for those autoantibodies that stimulate C3 convertase. 04:38 But if I really wanna make that biopsy or if I really wanna make that diagnosis pathologically, I'm going to need a renal biopsy. 04:47 So, unlike microscopy, it looks exactly like we look at with the immune complex mediated MPGN. 04:54 It's going to be hypercellular, lobular in appearance, and we may see some of that tram tracking or double contouring of the basement membrane. 05:02 But immunofluorescence in EM are very different. 05:05 Here is a patient who has dense deposit disease and you can see that C3 is stained throughout an intramembranous region. 05:13 On electron microscopy, this is probably one of the most beautiful electron microscopies that we have but this essentially shows intramembranous dense deposit, hence the name. 05:25 It's a ribbon-like appearance that's very dark that follows the entire glomerular basement membrane and that is very pathognomonic for dense deposit disease. 05:35 Now, I don't have a specific image for C3GN because it can manifest in a lot of different ways but typically on light microscopy, it will look hypercellular and lobular, and on immunofluorescence, we are only gonna see C3, and that is your clincher in giveaway diagnosis for C3GN. 05:54 So, how do our patients do who have complement-mediated MPGN? For dense deposit disease, there's about a 70% progression to end-stage renal disease at 9 years. 06:04 With C3GN, it's not very well-characterized but they probably have somewhat better of a prognosis than our patients with dense deposit disease. 06:15 In terms of treatment, if these patients have nephrotic syndrome, then they absolutely will need the non-specific therapy or treatment that we've discussed earlier with our nephrotic syndrome lectures and that includes ACE inhibitors, angiotensin receptor blockers, loop diuretics to move volume, a low-sodium diet, and statin therapy if those patients have hyperlipidemia. 06:35 Patients who have C3 nephritic factor, we can actually do plasma exchange and think about why we're doing that. 06:41 If that patient contains an auto -- or has an autoantibody in their serum, we can actually take that plasma, remove it, and replace it with either albumin or fresh frozen plasma. 06:52 In patients who have deficient factor H or factor I, those were those regulatory proteins that inhibit complement, then we can simply give them a plasma infusion that has the factor H and factor I in it. 07:06 Now, there is a very exciting treatment that is out there, eculizumab, and that is a monoclonal antibody to C5 protein. 07:15 And so, if you think about what's going on with alternative pathway of complement activation, remember that membrane attack complex is getting turned on by C3 degradation. 07:24 If we can have an antibody that inhibits that process like eculizumab, then there's hope for these patients. 07:31 It's not yet FDA approved for this particular disease process but it is something that we have in our armamentarium to treat our patients with either dense deposit disease or C3GN. 07:41 Part of the problem with our eculizumab is that it is quite costly and it can be on the order of $200,000 to $500,000 per year.
About the Lecture
The lecture Complement Mediated MPGN – Nephritic Syndrome by Amy Sussman, MD is from the course Nephritic Syndrome.
Included Quiz Questions
Which of the following statements is true regarding complement-mediated MPGN?
- It is caused by disorders in the alternative complement pathway.
- It is a disease of the elderly.
- The clinical manifestations are limited to the kidneys.
- It is associated with dysproteinemia.
Which of the following is associated with complement-mediated MPGN?
- Low C3 alone
- Low C3 and C4
- Elevated AH50
- Low CH50
- Elevated IgM and IgG levels
Which histopathologic changes are associated with complement-mediated MPGN?
- Electron microscopy reveals intramembranous ribbon-like deposits.
- Light microscopy reveals hypocellular lobulated glomeruli.
- Immunofluorescence microscopy reveals positive staining for C3 and immunoglobulins.
- Congo red stain shows C3 dense deposits.
Which of the following is involved in the pathogenesis of complement-mediated MPGN?
- Autoantibodies that stabilize C3 convertase
- Persistent HCV antigenemia
- Paraneoplastic glomerulopathy
- Autoantibodies that stabilize Factor H and Factor I
Author of lecture Complement Mediated MPGN – Nephritic Syndrome

Amy Sussman, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
