Playlist
Show Playlist
Hide Playlist
Sickle Cell Anemia: Hemoglobin Electrophoresis

-
Slides Sickle Cell.pdf
-
Reference List Pathology.pdf
-
Download Lecture Overview
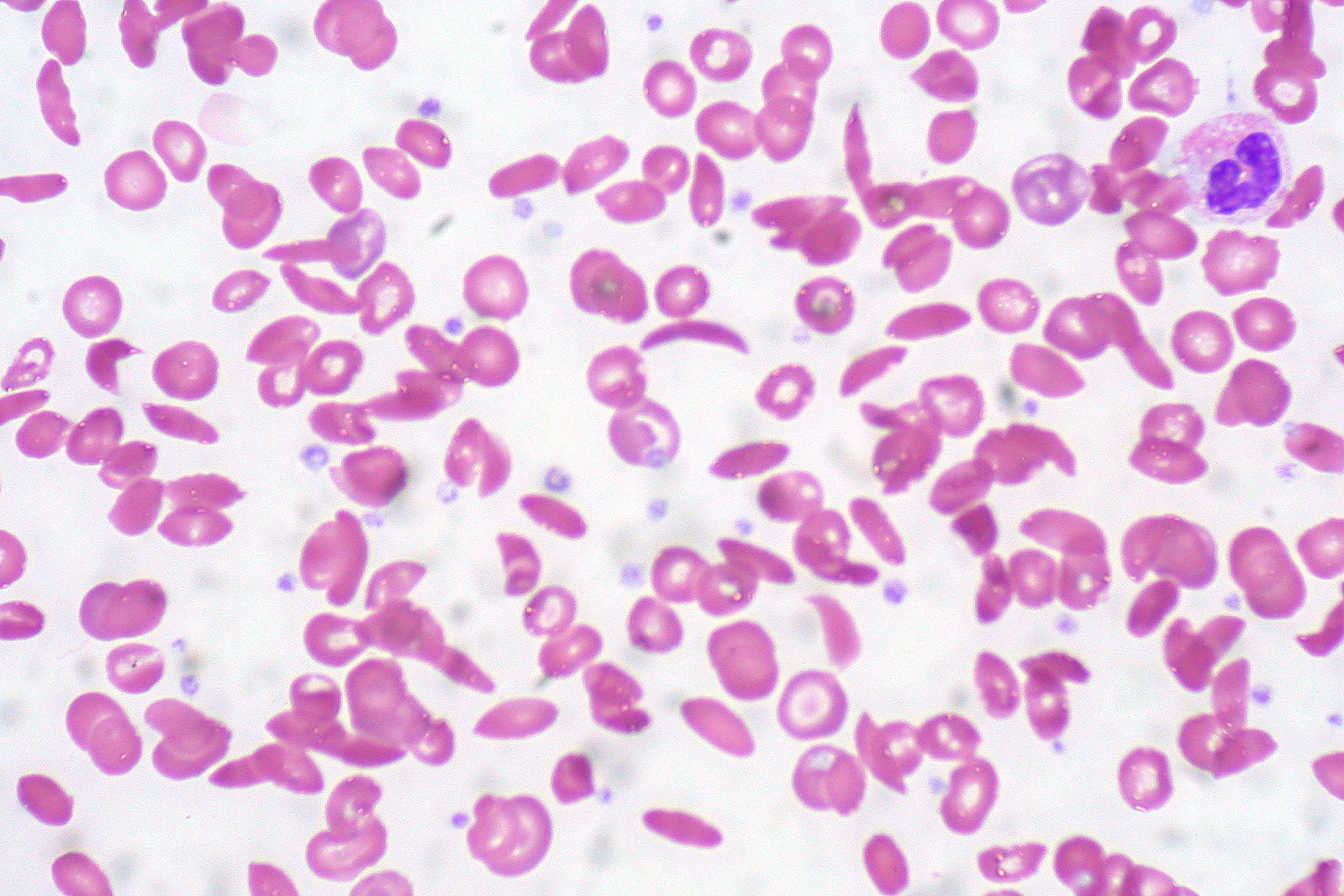
00:00 Quickly walk through trait and disease. Let's set this out for you. 00:06 This is still hemoglobin electrophoresis. On top, well, take a look at the following. 00:12 What is normal hemoglobin A? A whopping 97%. It's dropped down to 52%. 00:18 But you still have hemoglobin A. Take a look at hemoglobin S. It's at 45%. 00:24 So you tell me, which type of sickle pattern? Good. Sickle cell trait. One S. 00:31 So, it causes spontaneous sickling in peripheral blood. 00:34 And then it's still all normocytic hemolytic. 00:37 In the bottom picture, oh my goodness, no hemoglobin A. 00:40 Most of it is in the form of hemoglobin S. This is? Good. 00:46 Sickle cell disease homozygous. No hemoglobin A. 00:50 This is dangerous. Right? Really, really dangerous. 00:53 The spleen will be damaged. Hydroxyurea is what you're thinking about. 00:57 Because what does it do? It increases fetal hemoglobin. Fetal hemoglobin. 01:03 And that's what you wanna do in this condition because in sickle cell disease you have no hemoglobin A. 01:08 Maybe bone marrow transplantation if necessary because of reticulocytosis and at some point maybe parvovirus B19 kicks in. Fluid. Fluid. Fluid. 01:17 Analgesics. All part of your treatment regimen overall for sickle cell disease.
About the Lecture
The lecture Sickle Cell Anemia: Hemoglobin Electrophoresis by Carlo Raj, MD is from the course Hemolytic Anemia – Red Blood Cell Pathology (RBC).
Included Quiz Questions
Which of the following values represents a normal hemoglobin electrophoresis pattern?
- Hemoglobin A, 97%; hemoglobin A2, 2%; hemoglobin F, 1%
- Hemoglobin A, 90%; hemoglobin A2, 6%; hemoglobin F, 4%
- Hemoglobin A, 87%; hemoglobin A2, 12%; hemoglobin F, 1%
- Hemoglobin A, 90%; hemoglobin A2, 2%; hemoglobin F, 8%
- Hemoglobin A, 97%; hemoglobin A2, 1%; hemoglobin F, 2%
Author of lecture Sickle Cell Anemia: Hemoglobin Electrophoresis

Carlo Raj, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
