Playlist
Show Playlist
Hide Playlist
Pediatric Sickle Cell Disease

-
Slides Sicklecelldisease Pediatrics.pdf
-
Download Lecture Overview
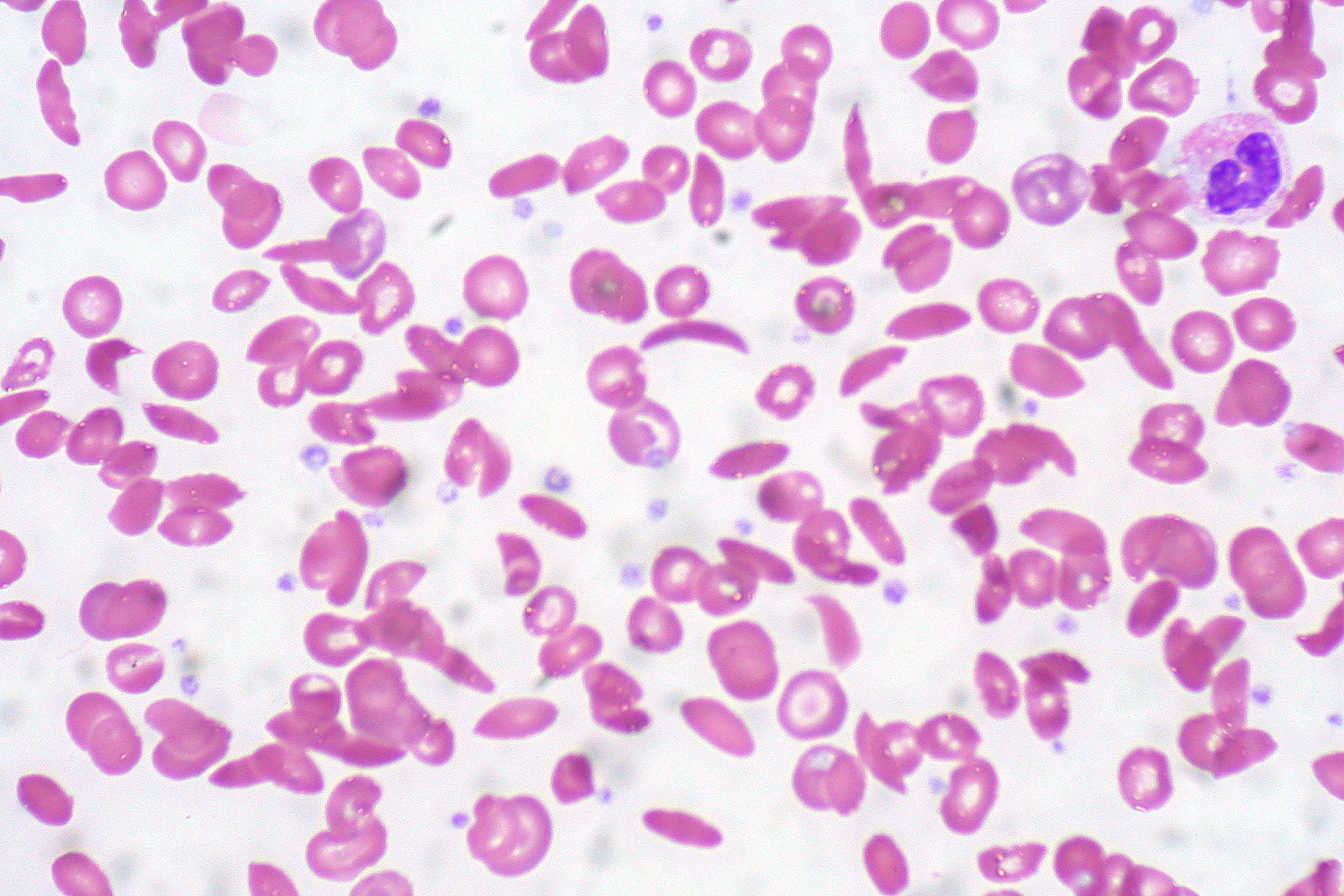
00:01 In this lecture, we will discuss sickle cell disease in children. 00:06 Let’s recall some basic science about sickle cell disease. 00:09 This is an autosomal recessive condition. 00:12 There is a point mutation for codon 6 on the beta-globin gene on chromosome 11 at position 15.5. 00:19 This point mutation results in a single change in amino acid which makes the hemoglobin less soluble. 00:27 We call a patient with the sickle gene hemoglobin S or HbS and a normal patient is HbA. 00:35 So a patient who is homozygotic for sickle cell disease will typically be hemoglobin SS and a normal patient will be hemoglobin AA and the carrier will be hemoglobin AS. 00:49 Okay. 00:51 Here is an example of cells that are sickling and you can see in the sickled cells labeled SC that these cells have less soluble hemoglobin resulting in that abnormal shape of the cell. 01:04 These cells are more friable and they don’t carry oxygen quite as well. 01:09 This is compared to the normal cells which are nice and round. 01:14 So the rate at which people have hemoglobin sickle genes is different depending on ethnicity. 01:21 And as you can see in the United States, almost 7%, a little bit over 7%, of African Americans carry a hemoglobin AS or have sickle trait. 01:32 This is compared to, for example, only 0.7% of Hispanic Americans. 01:37 It’s in 30% of patients from Sub-Saharan Africa. 01:41 In India, it’s around 13%. 01:44 In the Middle East, there’s a wide variability in terms of the rates, depending on where you are geographically. 01:50 In Greece, the rate is 1.5-7.5% and the Caribbean is 4-10%. 01:56 What’s interesting about these differences is that the disease or carrying the sickle gene seems to be somewhat protective of malaria. 02:07 So in populations that have evolved in areas where there is a lot of malaria, rates of sickle are higher. 02:15 So let’s talk a little bit about hemoglobin C. 02:19 So hemoglobin C is another mutation of the hemoglobin and it’s about a quarter as common as hemoglobin S. 02:28 In a patient who has one S and one C mutation, we call that hemoglobin SC disease. 02:34 Hemoglobin SC is not as severe as hemoglobin SS. 02:39 These patients have about half as many pain crises as the patients who are homozygotic for hemoglobin SS. 02:46 They have a lower risk of chronic issues. 02:49 They have a longer life expectancy by almost 20 years, about 65 versus 45. 02:55 And they have a higher incidence however of retinopathy in particular. 03:00 This is because they have a higher hematocrit which causes part of the problem. 03:06 Switching, there’s another variant that we should think about because thalassemia is also very common in areas where there is malaria and the patient may inherit a sickle gene as well as a thalassemia gene. 03:20 We call this sickle-beta-thal. 03:24 A patient may be beta zero or beta plus, if there is some hemoglobin A expressed. 03:31 So patients with beta zero expressed, almost no hemoglobin A and when they express some, they are beta plus. 03:38 So, sickled beta zero has a similar prognosis as hemoglobin SS disease because they are not making more hemoglobin A. 03:47 However, in patients with sickled beta plus, these patients have a better prognosis. 03:52 They are making more hemoglobin A and the hemoglobin is more soluble. 03:58 However, this is highly variable and it’s variable in terms of the course of illness depending on how much of their hemoglobin A is expressed. 04:08 Patients can also have sickle alpha thalassemia. 04:11 This is a milder illness than hemoglobin SS disease. 04:15 Another variant is patients with hemoglobin SS disease who have persistent fetal hemoglobin expression. 04:22 This is very protective against symptoms of sickle cell disease. 04:26 If a patient has a 10 or 20% persistence of hemoglobin F expression, they have a much better prognosis in terms of their sickle cell disease. 04:39 There are lots of other variations of sickle cell that are beyond the scope of this conversation, but just be aware that there are lots of variations and that sickle and thalassemia and even persistent fetal hemoglobin all interplay.
About the Lecture
The lecture Pediatric Sickle Cell Disease by Brian Alverson, MD is from the course Pediatric Hematology.
Included Quiz Questions
Which of the following is true regarding hemoglobin SC disease?
- Pain crises are less frequent than in hemoglobin SS disease.
- Retinopathy is less frequent than in hemoglobin SS disease.
- Life expectancy is less than that of hemoglobin SS disease.
- Anemia is severe.
- Splenomegaly is rare.
Which of the following is true regarding the pathophysiology of sickle cell disease?
- Point mutation in codon 6 of beta globin gene on chr 11
- Point mutation in codon 8 of beta globin gene on chr 11
- Point mutation in codon 6 of alpha globin gene on chr 11
- It is an autosomal dominant disease
- HbAS develops a full blown disease
Sickle cell trait is the least common in which population in the world?
- Hispanic Americans
- African Americans
- India
- Greece
- Caribbean
While comparing the patients and disease process of HbSC disease and HbSS disease, which of the following is true?
- HbSC disease: longer life expectancy
- HbSC disease has a higher risk of chronic issues
- HbSS: higher incidence of retinopathy
- HbSS: Fewer pain crises
- HbSC: Lower hematocrit
Author of lecture Pediatric Sickle Cell Disease

Brian Alverson, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
1 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
Excellent overview of the different types of the disease. It allows to wrap one's understanding around the types.
