Playlist
Show Playlist
Hide Playlist
Neuroblastoma in Children

-
Slides Neuroblastoma Pediatrics.pdf
-
Download Lecture Overview
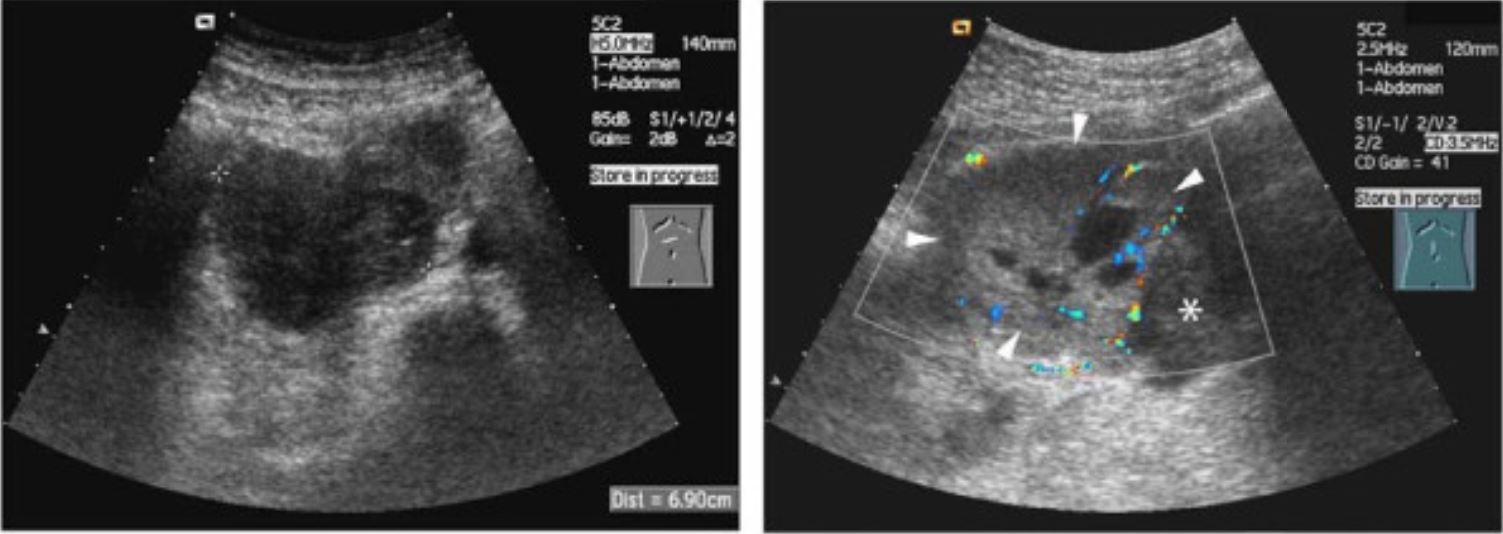
00:01 In this lecture, we will discuss neuroblastoma in children. 00:05 Neuroblastoma is a small blue round cell tumor. 00:09 It originates from the primitive neural crest cells and it's the most common primary site is in the adrenal gland, that's about 65% of cases. 00:19 But infants are more likely to have a cervical or a thoracic primary tumor. 00:25 It's most, and the most common malignancy in infants and it's the most common extracranial solid tumor of childhood. 00:34 So what is the basic epidemiology of neuroblastoma? Well, 90% of cases happen in kids under 5 years of age, the median age of diagnosis is 22 months of age, 15% of cancer-related deaths are children who die of neuroblastoma. 00:52 So, there is some genetics to it in that the N-myc protooncogene can be amplified in specifically tumor cells. 01:00 It's not an inherited gene. 01:02 It's what is the genetic error in patients with this disease and in those patients who have this mutation, they will have a worse prognosis and we use this genetic mutation to assess risk in children by looking for it in cancer cells. 01:18 This is also associated in patients with Hirschsprung's disease, with neurofibromatosis, and with Beckwith-Wiedemann syndrome. 01:27 So, to diagnose neuroblastoma, we have to do a history, a physical exam, and we're gonna do some imaging. 01:35 Let's start with the history. 01:36 Typically, these patients will present with abdominal mass. 01:40 That's especially true of the ones who have adrenal tumors. 01:43 They may have bleeding or bruising if the cells have invaded the bone marrow and causing a thrombocytopenia. 01:51 Additionally, occasionally it can develop a urinary obstruction from physical obstruction of urine outflow because of tumor cells. 01:59 Likewise, they may also have constipation. 02:02 Constitutionally, these patients may have lethargy, weakness, irritability, weight loss, or pain. These are all possible. 02:12 So, next we do a physical exam. 02:15 On exam, there are many particular symptoms which are specific to neurofibromatosis and I wanna walk through a few of them because some of them are fairly well known. 02:27 First of all, they may just have primary symptoms of the primary mass which is generally abdominal mass. 02:32 For liver metastasis, they may have a hepatomegaly. 02:36 If they have involvement of thoracic mass, they may develop Horner's syndrome which is the triad of miosis, ptosis, and anhidrosis. 02:47 If they have spinal cord compression, they may develop lower extremity weakness, urinary retention, and constipation, so that may also be from a neurologic impingement of functioning. 03:01 Patients may develop anisocoria and heterochromia in cervical masses. 03:08 Heterochromia is different colored eyes. 03:10 They may also develop blue, nontender, subcutaneous nodules if they have skin metastasis of these tumors, and they may develop raccoon eyes if they have orbit metastasis. 03:25 Lastly, there is a unique syndrome called opsoclonus myoclonus syndrome in patients with neuroblastoma. 03:34 This is a remarkable syndrome to see and is probably worth going to YouTube and putting that in and seeing if you can find a video. 03:42 These are jerking, rapid, involuntary eye movements associated with truncal and cerebellar ataxia and they are pathognomonic for pediatric neuroblastoma. 03:54 So, what is this opsoclonus myoclonus syndrome? Jerking, rapid, involuntary eye movements, truncal and cerebellar ataxia, it's an autoimmune reaction to the neuroblastoma cells, and these antibodies, these autoantibodies react against cells in the cerebellum causing the syndrome. 04:16 Lastly, we will do diagnostic testing. 04:20 So, one of the classic tests we will get will be in the urine where we check for urine catecholamines. 04:27 Typically, VMA and HVA which are vanillylmandelic acid and homovanillic acid are elevated in patients with neuroblastoma, so we will check a urine VMA and HVA. 04:41 The CBC may show signs of marrow infiltration but in low-grade disease, it might not. 04:48 Often we'll see an elevated ferritin which is really an acute phase reactant that can be elevated in the setting of neuroblastoma. 04:56 The diagnosis is confirmed by biopsy. 05:00 We find the tumor, we biopsy, we send it to the lab, and they will send us the pathology back and the histology back which will make the diagnosis. 05:08 We will also test that tumor for presence of the N-myc amplification gene, which confers a worse prognosis, and we will stage the bone marrow for biopsy. 05:18 We'll biopsy it to look and see if the bone marrow is involved. 05:22 We will also do an MRI scan of the primary tumor to determine the extent of disease. 05:29 Here's a patient with an adnexal tumor and you can see that that's a pretty large tumor and they wanna figure out how involved it is. 05:37 We will do CT of the chest, abdomen and pelvis, to look for where there might be metastasis and we may do a bone scan to look for metastasis as well. 05:47 So here's an example of a bone scan. 05:49 You inject the MIBG radionuclide and it's an isotope that's preferentially taken up in catecholamine-producing cells such as neuroblastoma. 06:01 90% of neuroblastoma cells will take up this isotope, and here's a patient you can see with many bony metastases of disease. 06:09 So then we stage the disease, and this is based on whether it's localized or disseminated. 06:16 That doesn't necessarily confer the entirety of prognosis. 06:22 There is a variety of type, stage IV disease for example, which actually carries a more beneficial likelihood of survival. 06:31 So then we will use histology and presence of the N-myc amplification and we'll look for DNA ploidy of the tumor. 06:39 We'll look for chromosomal alterations in the tumor, and we'll look at the age of the patient, and all these things together determine the risk of this patient's disease. 06:48 For high-risk patients, we're gonna treat these patients very aggressively. 06:54 They will get intensive chemotherapy, surgical resection, an autologous stem cell transplant, radiation, and immunotherapy. 07:04 All of these things are potential tools against very high-risk patients. 07:07 High-risk patients confer a high risk for death from this disease, so we definitely are very aggressive with them. 07:15 For low-risk patients, we often do surgical resection, we'll do a less intensive chemotherapy, and then we will observe them for a period of time and see how things are going and hope that that may be sufficient. 07:28 This is that stage IV subset who actually have a favorable prognosis. 07:35 They're usually under a year of age. 07:37 They have a localized primary tumor and are stage I or II, they have metastases that are limited to the skin, the liver, and the bone marrow, and they have no poor prognostic indicators in terms of mutations. 07:50 These patients actually have a pretty favorable prognosis. 07:53 But generally, the low-risk patients have a good prognosis, the high-risk patients usually more than half of the time will die despite aggressive therapy. 08:05 And we do incorporate immunotherapy for high-risk patients and that has been shown to dramatically improve prognosis for neuroblastoma patients. 08:15 So, that's my brief review of neuroblastoma in children. 08:20 Thanks for your time.
About the Lecture
The lecture Neuroblastoma in Children by Brian Alverson, MD is from the course Pediatric Oncology. It contains the following chapters:
- Neuroblastoma in Children
- Location of Metastases and Clinical Presentation
- Risk Determination
Included Quiz Questions
Neuroblastoma most likely originates from which of the following?
- Neural crest cells
- Cerebellar cells
- Lymphatic tissue
- Adrenal cortex cells
- Fibroblasts
Neuroblastoma is NOT associated with which of the following?
- Panhypopituitarism
- Hirschsprung’s disease
- Neurofibromatosis
- Beckwith-Wiedemann syndrome
- N-myc proto-oncogene mutation
Which of the following types of pediatric tumors is known to be a common cause of the paraneoplastic opsoclonus myoclonus syndrome?
- Neuroblastoma
- Medulloblastoma
- Ependymoma
- Astrocytoma
- Glioma
Which of the following confirms a diagnosis of neuroblastoma?
- Tumor biopsy
- Urine catecholamines
- Complete blood count with differential
- Serum ferritin
- Bone marrow biopsy
Author of lecture Neuroblastoma in Children

Brian Alverson, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
1 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
Excellent lecture as usual. A complex topic made easy. Thanks a lot!
