Playlist
Show Playlist
Hide Playlist
Interstitial Lung Disease (ILD): Pathology

-
Slides 06 ILD InterstitialLungDisease RespiratoryAdvanced.pdf
-
Download Lecture Overview
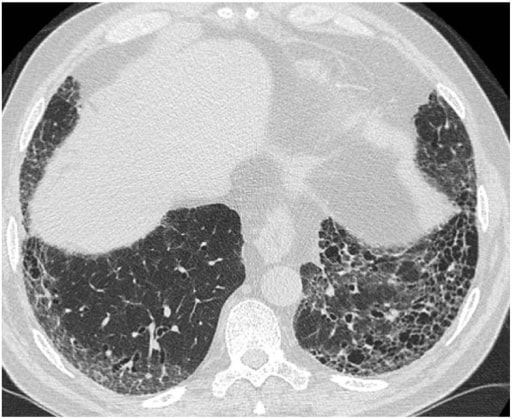
00:00 It is the pathology of the infiltration of the alveoli and interstitium that dictates what ILD the patient has. For pulmonary fibrosis, there are two main patterns. 00:10 One is a largely inflammatory cell infiltrate into the alveoli and interstitium, and that’s called non-specific interstitium pneumonitis or NSIP. 00:19 Another is where you have large amount of increased extracellular matrix, collagen, and fibroblast, a sort of fibroblastic infiltration of the lungs leading to essentially what looks like scar tissue forming in the lung and that is called Usual Interstitial Pneumonitis, (UIP). It is not entirely clear, but probably, NSIP may evolve into UIP in some patients. 00:46 However, with other interstitial lung disease, you get a completely different pattern of lung infiltrations, for example, sarcoidosis as mentioned already. You get infiltration of non-caseating granulomas, and hypersensitivity pneumonitis, and also you get non-caseating granulomas but much more poorly formed than you do in sarcoid, and associated with mononuclear infiltrate and a peribronchial fibrosis leading to some small airways obstruction. 01:07 And then diseases such as pulmonary eosinophilia and lymphocytic interstitial pneumonitis cause respectively as the names might suggest an eosinophinic and a lymphocytic infiltration of the lung. So the pathology of the ILD is dictated by the underlying cause.
About the Lecture
The lecture Interstitial Lung Disease (ILD): Pathology by Jeremy Brown, PhD, MRCP(UK), MBBS is from the course Interstitial Lung Disease (ILD).
Included Quiz Questions
Which of the following pathological patterns are NOT related to interstitial lung disease?
- Caseating granulomas
- Usual interstitial pneumonitis
- Nonspecific interstitial pneumonitis
- Noncaseating granulomas
- Eosinophilic consolidations
Which interstitial lung disease will most likely have poorly formed noncaseating granulomas in the lungs?
- Hypersensitivity pneumonitis
- Collagen vascular disease
- Pulmonary eosinophilic pneumonitis
- Lymphocytic interstitial pneumonia
Which of the following statements about interstitial lung disease is TRUE?
- Interstitial lung diseases are characterized by noninfectious infiltration of the interstitium.
- Interstitial lung diseases usually do not affect the transfer factor.
- Interstitial lung diseases can usually be diagnosed accurately using a chest X-ray.
- Interstitial lung diseases usually cause obstructive defects seen on spirometry.
Author of lecture Interstitial Lung Disease (ILD): Pathology

Jeremy Brown, PhD, MRCP(UK), MBBS
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
