Playlist
Show Playlist
Hide Playlist
Expansions and Huntington's Disease

-
Slides 03 Expansions and Mitochondrial Inheritance SingleGeneDisorders.pdf
-
Download Lecture Overview
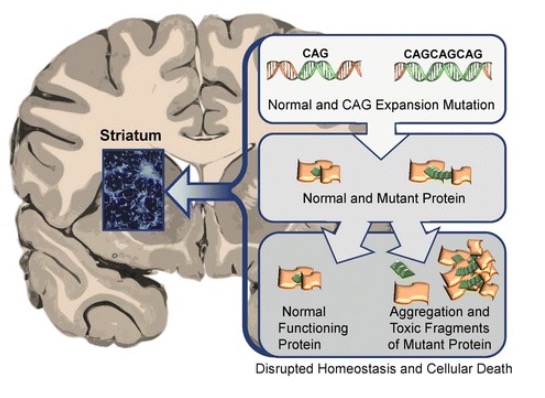
00:01 In our final lecture on single gene inheritance, we’ll be looking at expansions and mitochondrial inheritance. 00:10 Now, what are expansions? I brought them up very briefly in an earlier lecture. Trinucleotide repeat expansions are what we’re talking about. If you don’t recall anything about expansions previously, you can probably get from the title trinucleotide, three nucleotides and repeat, they keep repeating and they keep expanding. These are dynamic mutations. They change from generation to generation. 00:42 As it’s passed down from one parent to their offspring, the number of repeats almost generally will increase. 00:52 We don’t really notice it when it’s decreasing. They are expanding repeats. The repeats are normal in the population, regular number. At a certain number, they become abnormal and result in some phenotypic problems. Again, they’re expanding as they go from generation to generation. You can recall very easily from the title exactly what’s going on because they’re named appropriately. There are two main diseases that we’ll be considering: Huntington’s disease which is a polyglutamine disease, meaning we get many more glutamine residues in a particular gene than we would like to have. 01:38 Then Fragile X, I brought that up when we were talking about X-linked disorders because indeed, it is an X-linked disorder. Let’s take a look first at Huntington’s disease, a polyglutamine disease. 01:55 It’s an expanding repeat. It’s an expanding repeat of C, A, and G, so CAG, CAG, CAG. It’s normal to have about 25 repeats of this in the Huntington gene which is on chromosome 4. It’s not a sex-linked disease. 02:14 This is an autosomally inherited disease. The CAG from uneven crossing over or from maybe translocations but either way, the CAG repeat, replication causes these extra repeats. Once an individual has more than 40 repeats, we see expression of Huntington’s disease. What the repeats end up doing is having multiple glutamines added to the chain. Those glutamines end up forming a dysfunctional Huntington protein. 02:55 We’ll talk a lot more about the manifestations of Huntington’s disease in the aging lectures where we’ll look at disorders of aging. But for here, we’ll just look at this expanding CAG repeat causing multiple glutamines. Now, anticipation is the concept of having the disease display progressively earlier and earlier in the family line. The longer the repeat, the earlier the disease is expressed. 03:38 So, it is anticipated from generation to generation. We also note a paternal transmission bias. 03:45 If an individual has more than 25 but less than 40, we call this a premutation. That premutation is called a premutation because it can continue to expand as it is passed down from generation to generation. 04:03 A paternal transmission bias means that it is transmitted and elongated more through the paternal line than it is through the maternal line. A father that has a premutation could pass on an allele that would become mutated in the next generation. Not that it couldn’t happen from a female parent but it is biased towards the male parent having the premutation turn into a full-on mutation. Again, that’s when it reaches about 40 different sequential CAG repeats. Too many glutamines causes dysfunctional protein. 04:47 Here is a pedigree showing Huntington’s disease. Below it, you can see a Southern blot analysis. 04:56 Recall that in Southern blotting, as we talked about in molecular genetics, involves moving DNA through a gel and then making a radioactive print of that. The further through the gel that the fragments move, the shorter the pieces because shorter things are going to go through faster. Here you can see that the individual number 2 all the way over on the right in the first generation homozygous has only one length fragment for the normal length of CAG repeats which is about 25. You can also see that for individual 3. Now, you’ll see noted beneath each individual the number of repeats that they have. 05:50 For a moment, knowing what you know that the larger the repeat, the earlier the condition shows, which individual in this pedigree is likely to display Huntington’s disease at an earlier age? Okay, you’re right. The one with 103 below it, generation 2, individual number 5 is going to have the earliest onset and perhaps the most severe symptoms of Huntington’s disease. That’s what we call the age dependent penetrance. As note at the time of this pedigree, individual with 42 repeats at age 50 did not yet display Huntington’s but it’s expected because he has more than 40 that he will display the symptoms of Huntington’s later in life. Take a look at this figure and think for a moment what is it showing. It’s really a great figure because it summarizes everything that I’ve talked about with the polyglutamine disease of Huntington’s. You can see on the X axis, we’re talking about the number of CAG repeats and age and what’s going on in this blue area. Why don’t we see anything all the way over here on the left side? We don’t see anything because we need to have 40 repeats. Once we have 40 repeats or around 40, we may have expression. You know that the more repeats we have, the earlier the age of expression. This graph does a really nice job of summarizing the concepts that we’ve learned in Huntington’s disease. Good one to keep in mind.
About the Lecture
The lecture Expansions and Huntington's Disease by Georgina Cornwall, PhD is from the course Single-Gene Disorders. It contains the following chapters:
- Expansions and Mitochondrial Inheritance
- Polyglutamine Disease - Huntingston's
Included Quiz Questions
Which of the following regarding genetic disorders of trinucleotide repeats is most accurate?
- They may increase in number during intergenerational transmission.
- They usually manifest as a disease when the number decreases below a threshold.
- They are always X-linked.
- They accumulate missense mutations.
- The dominant or recessive inheritance pattern of disease transmission does not apply to these kinds of disorders.
Which amino acid is repeated excessively in a chain of huntingtin protein?
- Glutamine
- Glycine
- Asparagine
- Lysine
- Alanine
What is the trinucleotide sequence repeated in Huntington’s disease?
- Cytosine-adenine-guanine
- Cytosine-guanine-guanine
- Cytosine-thymine-guanine
- Adenine-thymine-guanine
- Guanine-adenine-adenine
Which of the following terms describes a premutation that is expanded more frequently through the father's cell line?
- Paternal transmission bias
- Maternal transmission bias
- Y chromosome transmission bias
- Generational transmission bias
- X chromosome transmission bias
How do increasing numbers of trinucleotide repeats in Huntington's disease affect disease presentation?
- The disease presents at an earlier age.
- The disease presents at a later age.
- The disease presents more frequently in males.
- The disease presents in more offspring.
- The disease presents with milder symptoms.
Author of lecture Expansions and Huntington's Disease

Georgina Cornwall, PhD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
