Playlist
Show Playlist
Hide Playlist
Atherosclerosis: Diagnosis and Treatment

-
Slides Pathophysiologic Approaches.pdf
-
Reference List Pathology.pdf
-
Download Lecture Overview
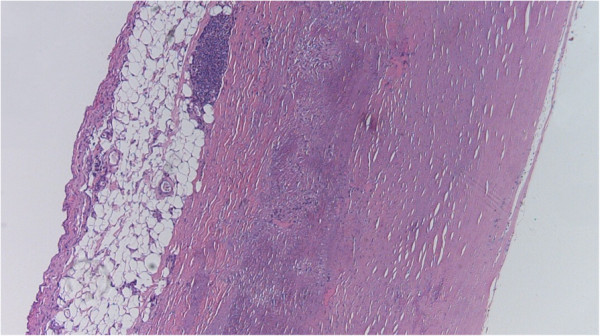
00:01 Okay, then we're coming up on the final two sessions in this block on atherosclerosis, and try to think about ways that we might be able to diagnose and treat patients. 00:16 With that, here's where we are in our roadmap. 00:19 We've covered risk factors, how the plaques are formed, the impacts of plaque morphology, and the complications when plaques go bad. 00:27 And now into the approaches for the diagnosis and treatment. 00:33 Okay, so we have talked previously, about athero-prone areas of the vasculature and atheroprotected areas of the vasculature. 00:43 And in an earlier session, we talked about the internal and external carotids in where there was laminar flow, and where there wasn't laminar flow. 00:52 And a number of groups have looked at how does that flow characteristic, somehow translate information to the endothelial cell, so that it behaves differently and it protects against atherosclerosis, or it is more prone to atherosclerosis. 01:14 And the groups that have worked on this have taken the different waveforms and subjected the endothelial cells to those shear stresses in mechanical setups. 01:28 And one of the major factors that popped out of this analysis is Kruppel-like factor two, KLF-2, and I'll just say KLF2. 01:38 KLF2 is very highly expressed on the blue bar, or the blue column, that it's atheroprotective. 01:49 And it's expressive, very, very low levels in athero-prone treated waveform endothelial cells. 01:58 So what is KLF2, Kruppel-like factor 2? It's a zinc finger transcription factor so it's something that's going to be important for regulating DNA transcription. 02:08 And it's something that is upregulated by laminar shear in atheroprotected portions of the vasculature. 02:16 How does this happen? Well, some of the steps aren't known. 02:19 So we do know that atheroprotective flow through some sort of mechanical transduction mechanism yet undiscovered that's up to you, will then lead to the activation of a number of intracellular kinases and they have these names. 02:35 It's not important that you remember these names, only that there is this upregulation and we get more KLF2 when we have atheroprotective flow. 02:44 That's great, a lot of good things will happen by having that KLF2 transcription factor synthesized. 02:52 Now, the really interesting part that we had no idea when we developed the drug statins influenced this, what you're saying how does that happen? Well, see that HMG-COA reductase, which is the first rate limiting step in the synthesis of cholesterol will drive subsequent downstream production of things like mevalonate. 03:11 Mevalonic acid will inhibit one of the upstream kinases, MEKK3, if you care, and that will inhibit the production of KLF2. 03:25 So when we are actively synthesizing a lot of cholesterol, we're inhibiting KLF2. 03:30 If I inhibit cholesterol synthesis with statin, I block that I take away that block, a MEKK3, and I get upregulated KLF2. 03:40 So atheroprotective flow, and a drug that we designed for completely unrelated reasons, converge on the production of this really great factor, KLF2. 03:52 So KLF2, what is it doing? So it's anti inflammatory, it tunes down the inflammatory response in endothelial cells, and in inflammatory cells, you get improved cholesterol metabolism, that is to say you reduce cholesterol as a result of KLF2. 04:11 You get reduced vascular adhesion so you make, you recruit fewer inflammatory cells, which are going to be the early driving steps in the development of atherosclerosis. 04:22 And you have reduced coagulation. 04:24 So as I say, a lot of terrific things happen when you get upregulation of KLF2 and we do it naturally in many areas in our in our vasculature. 04:35 And we can do it artificially, therapeutically by administering statins. 04:41 So again, it may be another reason that it should be in the water supply. 04:44 Just joking. 04:46 Okay. So how do we know which atherosclerotic plaque is more likely to rupture? We've talked about this in the previous session. 04:52 And the big answer is, we don't know. 04:55 We don't have reliable imaging or markers to identify which plaque is going to rupture and this is just an image to show you an atherosclerotic plaque, most of that is yellow and green, and then that big, red thing in the middle, that's thrombus that occurred when the plaque ruptured just below that. 05:16 So, how do we know that? So how do we modify that at least? and plaque stability and vulnerability is driven by a number of factors including inflammatory risk. 05:29 So we can monitor CRP as a marker, C-reactive protein as a marker for inflammation. 05:36 And we can modulate some of that inflammatory risk by eliminating, treating inflammatory diseases, such as autoimmune diseases like rheumatoid arthritis, or lupus erythematosus. 05:50 We can modulate hopefully inflammatory cell recruitment and activation. 05:55 Those cells are going to elaborate cytokines and growth factors but things like statins will reduce that. 06:02 Angiogenesis and neovascularisation will also impact whether or not a plaque will rupture. 06:07 The greater the vascularization, the more likely the plaque will be vulnerable. 06:14 The recruitment and activation of smooth muscle cell precursors and their proliferation are also going to be important and that can also be driven by things like statins. 06:24 And then finally, it's the Yin and Yang. 06:26 It's the push and pull of matrix synthesis, and matrix remodeling. 06:31 And going forward thinking about how we can stabilize plaque by administering drugs that limit matrix remodeling. 06:41 Statins do that to a certain degree but not entirely. 06:47 And with that, we've finished this very short chapter on pathogenesis and how we can modify plaque.
About the Lecture
The lecture Atherosclerosis: Diagnosis and Treatment by Richard Mitchell, MD, PhD is from the course Atherosclerosis.
Included Quiz Questions
Kruppel-like factor-2 (KLF2) expression is increased in what pattern of the waveform?
- Athero-protective
- Athero-prone
- Athero-static
- PR prolongation
- QT prolongation
Upregulation of KLF2 results in…
- …improved cholesterol metabolism.
- …pro-inflammatory cytokine production.
- …increased vascular adhesion.
- …increased coagulation.
- …turbulent arterial flow.
How do statins help in maintaining plaque stability?
- Reduce inflammatory cell recruitment
- Reduce platelet adhesion
- Activate C-reactive protein
- Promote matrix degradation
- Increase circulating precursors
Author of lecture Atherosclerosis: Diagnosis and Treatment

Richard Mitchell, MD, PhD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
